The first decision: amplicon sequencing or wgs? Or… both? :O
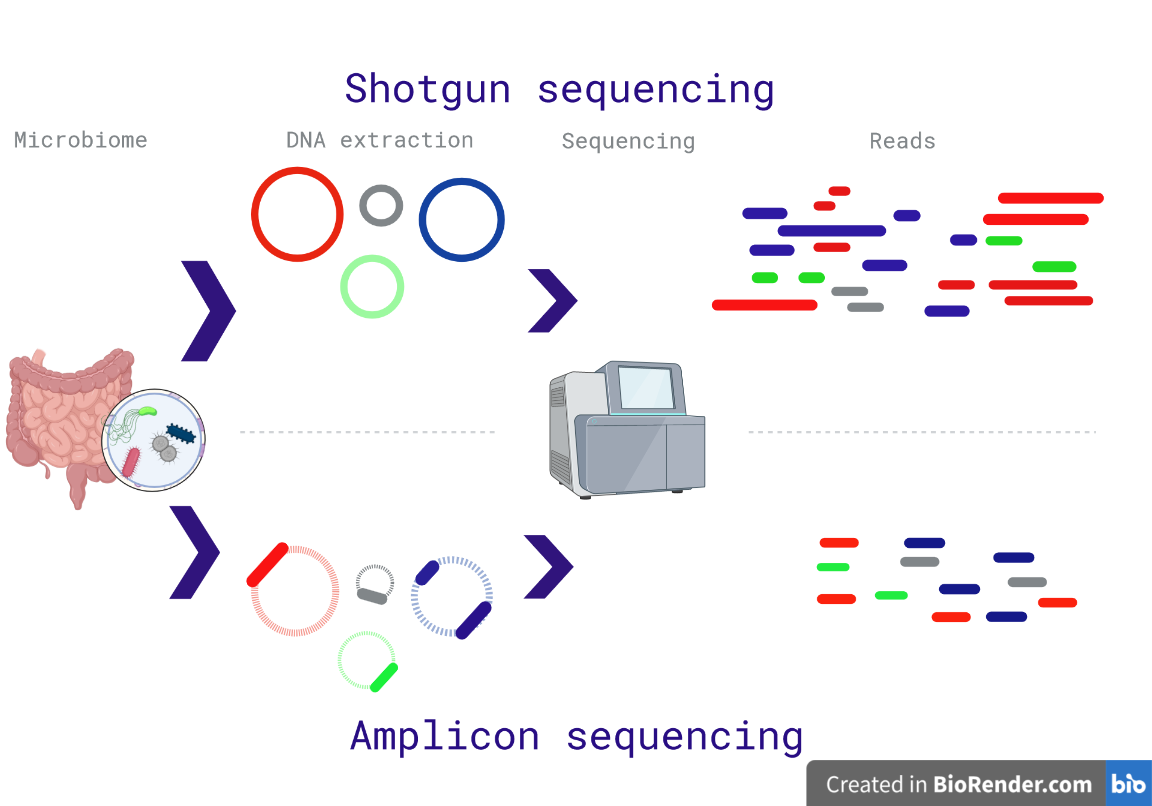
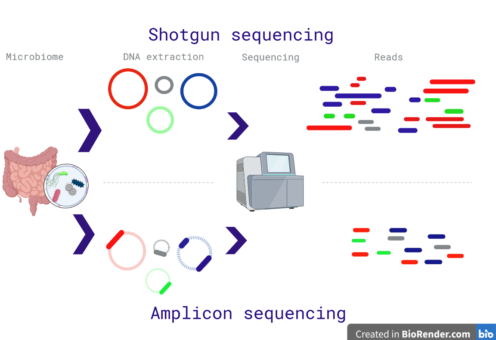
The microbiome pipelines can be divided in two categories, whole genome shotgun sequencing, and amplicon sequencing (Fig. 1).
Fig. 1: Differences between the shotgun and amplicon sequencing. The shotgun method sequences the entire DNA in a sample; while the amplicon method sequences specific regions. Red sentences represent cons; while green sentences are advantages. Created with biorender.
As suggested by the name, the shotgun approach sequence all the DNA in a metagenomic sample. In a few words, the DNA sample is randomly fragmented and amplified, after sequencing the fragments, the reads are assembled to recreate the DNA genomes (Fig. 1a).
On the other hand, the amplicon sequencing uses oligonucleotide probes to amplify specific sequences (a footprint for taxonomy classification). In microbiome research, the 16S is used for prokaryotes, 18S for eukaryotic microbes, and ITS (Internal transcribed spacer) for fungi.
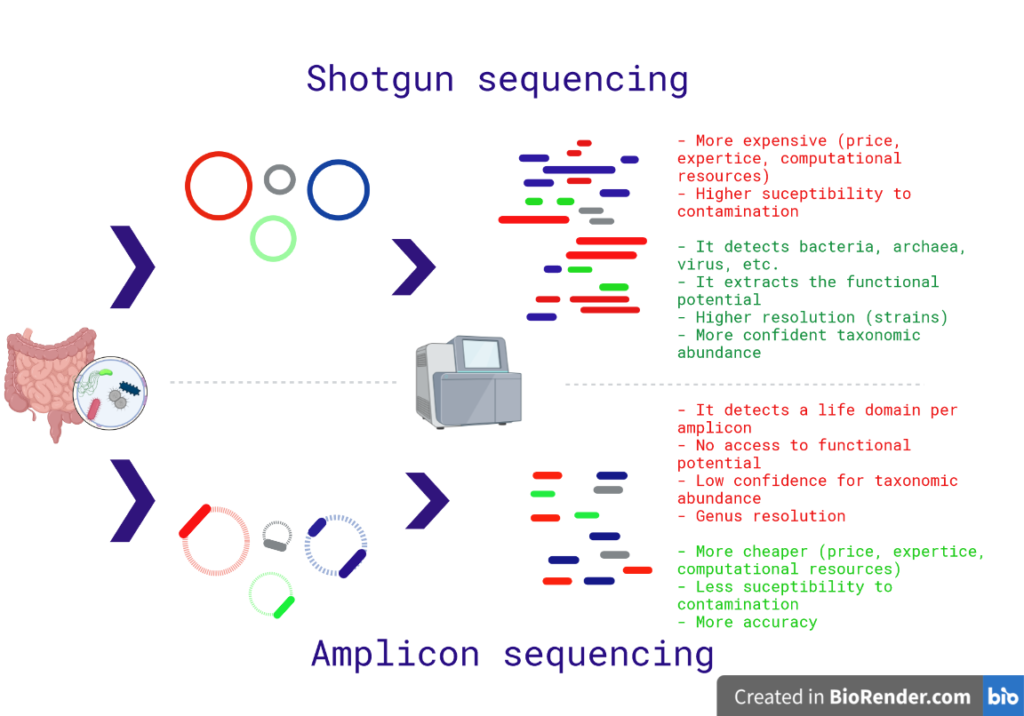
The preferred pipeline for taxonomy profiling is amplicon sequencing.
- It does not require to sequence and analyse the entire microbiome; therefore, it is cheaper.
- The conserved ribosomal regions allow to filter the host DNA. If you use a 16S oligonucleotide probe in a gut microbiome sample, the human DNA will not be amplified. Therefore, it has less susceptibility to contamination.
- It is more precise (it has less false positive classifications). First, the amplicon databases are more extensive and well curated. Second, the probability of labelling a random coding gene as a ribosomal one is low. The shotgun approach uses marker genes, proteins exclusive to a clade to make the classification. However, these marker genes can be classified as exclusive to a clade only by lack of information.
- It requires less bioinformatic expertise.
- It reports two life domains per amplicon at most. To classify bacterial, fungi, archaeal and eukaryotic microbes, you would need to make three assays: 16S, 18S, and ITS.
- The amplicon sequencing ignores any other gene, the functional potential of your samples will be ignored. With the amplicon sequencing, you can make raw speculations about the functional profiling of your samples based in the annotated genes of your identified taxa; however, the gene counts are completely inaccessible.
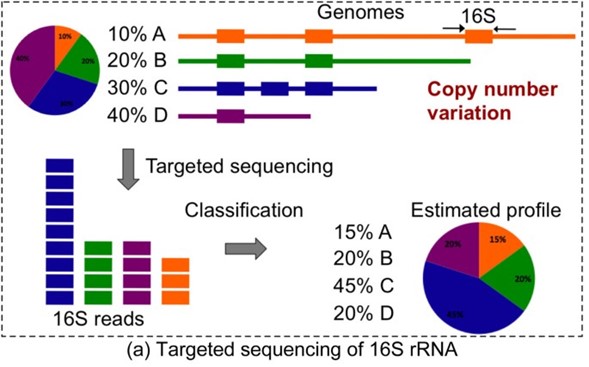
- A criticism to the amplicon sequencing is the low confidence for taxonomic abundance estimation. The amplicon sequencing is highly susceptible to PCR bias, the same oligonucleotide probe can be more effective to detect a Bacillus than a Nitrospirae. Fig. 3 shows the gene copy-number problem in amplicon sequencing. Acuracy
Fig. 2: The amplicon sequencing can overestimate a taxa abundance as a consecuence multiple 16S copies. Source
The gold standard for potential functional profiling is the shotgun approach
- The shotgun approach looks to sequence the entire genome of every Bug in the sample, it is more expensive (money, computational resources, expertise).
- It does not filter by life domain, it detects bacteria, archaea, viruses, etc.
- The shotgun approach access to the functional POTENTIAL in the microbiome. You must remember that many genes remain unexpressed under different conditions.
- It recovers single nucleotide variants (more resolution, at the level of strains).
- You can cross-reference the functional profiling with the taxonomic profiling (more details below).
- It makes better predictions of the taxonomic abundance.
- The shotgun databases encompass less species; therefore, it is more likely to deplete the taxonomic calling.
The same sample can display different results based in the used pipeline and database. Each approach answers different questions at different resolutions; therefore, you must identify your research interest and adapt your assays to the question. However, your budget, DNA host contamination, expertise and computational resources must be considered; if you have limited resources, you may need to adapt your research question to the available resources.
- If you are interested in confident taxonomic profiles, the amplicon pipeline may be your best option. Large numbers of samples can increase your preference for this approach. Besides, if you are particularly interested in species present in a group of samples and excluded from another rather than differential abundance, the amplicon sequence can be a better option. For example, in the identification of microbial footprints for sick plants in comparison with healthy plants, the precision is more important than abundance.
- If the sample belongs to a complex or rare environment, an amplicon database will be more suitable. For example, if you want to analyse sea samples, a shotgun database is more biased towards model organisms/environments.
- If you are interested in trace the taxonomic profiles at strain level, you may have to use the shotgun approaches.
- If your main interest is the functional profiling, you should use shotgun assays.
There is a lot of information about these pipelines. I recommend https://www.cd-genomics.com/microbioseq/amplicon-based-next-generation-sequencing-vs-metagenomic-shotgun-sequencing.html, https://blog.microbiomeinsights.com/16s-rrna-sequencing-vs-shotgun-metagenomic-sequencing, and https://astrobiomike.github.io/misc/amplicon_and_metagen.