I choose to work with a shotgun approach; Now, what? 😐
Fig. 3. Overview of a shotgun pipeline. After some quality filters to the metagenome reads, you can make taxonomic predictions before the assembly, or you can make taxonomic predictions with an assembled metagenome. Since the shotgun aproach is more suceptible to contammination, I would add a step: remotion of reads that map to human/host genome.
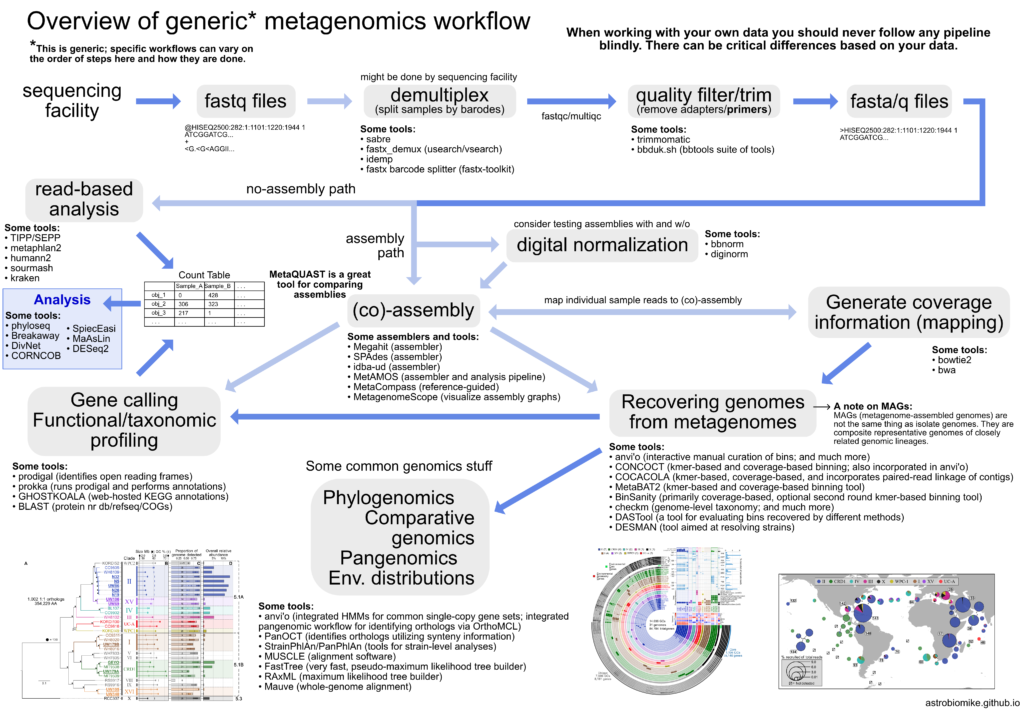
FIg. 3 describes the generic pipeline for shotgun sequencing analysis. I will talk about the phylogenetic classification.
Once you have your high-quality reads per sample, you have two options, make taxonomic profiling with the reads, or make taxonomical predictions with the assembled microbiome.
It is recommended to make taxonomic classifications directly from the reads. The reads are more confident representations of the DNA content in the microbiome; an assembly has corrected basepairs, insertions, deletions, etc. Furthermore, an assembly dismisses the predicted counts of species; repeated reads/sequences (that maybe belongs to different individuals of the same specie) are condensed into a representative one. Obtain confident abundance profiles with assembly classifications is more challenging.
The principle for any taxonomy classification is the comparison of the microbiome sequences properties against microbial reference genomes. There are two main classes of taxonomic classification programs: homology-based algorithms and composition-based programs.
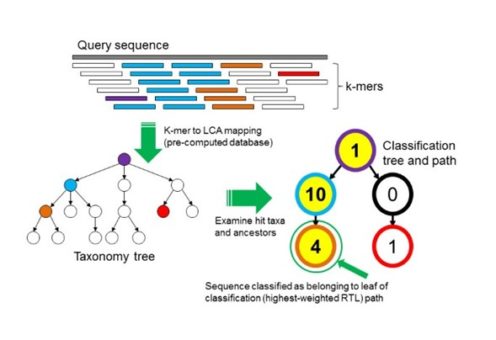
The most frequent method is the homology-based approach, which are divided into DNA, protein, and marker-genes algorithms. These methods have shown better performance in the analysis of fragments shorter than 1 kb; the metagenome reads are the common input. Their principle is the alignment of the microbiome sequences against a database composed of microbial reference genomes. Technically, you could use programs like diamond to align your microbiome sequences against a database with all the available microbial genomes. However, the increasing amount of reference genomes will delay your analysis. To overcome the problem, some programs have developed heuristic solutions, these solutions include the use of k-mers. For example, Kraken is a k-mer based classifier that uses reads and DNA databases (Fig. 4). It creates an index with the k-mers in a set of reference genomes (database), then identifies which k-mers are in the microbiome sequences (query) and creates a taxonomy tree. Finally, Kraken identifies the lower common ancestor (LCA) of the k-mers shared in the query and the database sequences; the N node with more accumulate k-mers starting in the LCA and ending in the N node is identified as the taxonomy classification of that query sequence.
Fig. 4: Kraken K-mer based approach. “To classify a sequence, each k-mer in the sequence is mapped to the lowest common ancestor (LCA) of the genomes that contain that k-mer in a database. The taxa associated with the sequence’s k-mers, as well as the taxa’s ancestors, form a pruned subtree of the general taxonomy tree, which is used for classification. In the classification tree, each node has a weight equal to the number of k-mers in the sequence associated with the node’s taxon. Each root-to-leaf (RTL) path in the classification tree is scored by adding all weights in the path, and the maximal RTL path in the classification tree is the classification path (nodes highlighted in yellow). The leaf of this classification path (the orange, leftmost leaf in the classification tree) is the classification used for the query sequence.” Source
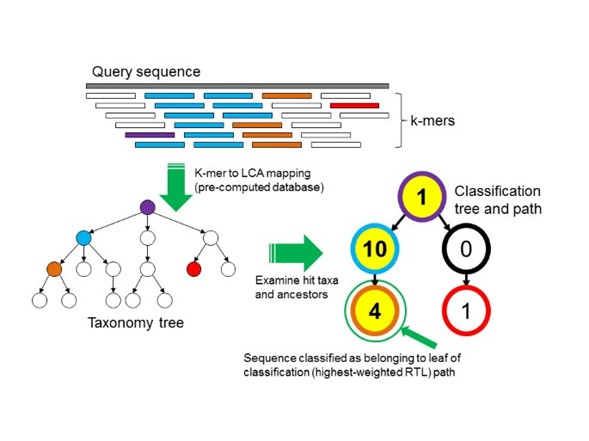
The k-mer size has a huge impact in your classification. Short k-mer length will be more flexible; therefore, the precision will be reduced (more false positives). A short-kmer length may be more suitable if you suspect that your sample has higher evolution rates (e.g., it is subjected to selective pressures) or, if your microbiota is composed of distant taxa (e.g., due to high genetic flux in a sea sample). On the other hand, longer k-mer sizes are more restrictive; a few random mutations can provoke you metagenomic sequence to be dismissed. Long k-mer sizes are preferred if you suspect that your samples are less diverse.
Another k-mer homology-based program is CLARK, which has a similar principle than Kraken. CLARK can classify DNA/RNA reads/scaffolds/contigs/etc. It makes an index per taxonomic rank; therefore, if you are interested in a specific taxonomic rank, Clark can reduce the memory requirements and time consumption.
Kraken and CLARK are DNA classifiers; these programs use DNA reference genome databases. These methods are more restrictive than the protein programs; every mutation affects the similarity between sequences. On the other hand, the protein sequences are more conserved despite the DNA mutations. Two facts must be considered, first, the current databases represent only a small fraction of the real microbial diversity. Besides this fraction is biased, it does not include many environments of interest for the microbiome research (extreme environments, complex communities, etc.). Second, the microbial word (especially the bacterial and virus world) has a high evolution rate. The mutational rate, and the taxonomy bias in the incomplete databases makes tricky to ask very strict hits, the DNA algorithms are stricter. And the microbial world has high density of protein-coding sequences; therefore, a protein approach for taxonomy classification may improve accuracy. Finally, using homology-based programs that classify reads based on a protein database has another advantage. If you make protein predictions of your metagenomic sample, you can cross reference the reads with a predicted protein with the taxonomy classification of this read; and repeat for each protein. Not every protein will have a taxonomy assignment; however, the cross references will be a hit of the metabolic redundancy in the sample. How many taxa has a predicted protein of interest? The higher abundance taxa, Hoe diverse (in functional potential) is it?
Fig. 5: MEGAN algorithm. Reads are the input data in MEGAN. Prior sequence alignment is required. Then, MEGAN is used filter the taxonomy classifications based in the LCA.
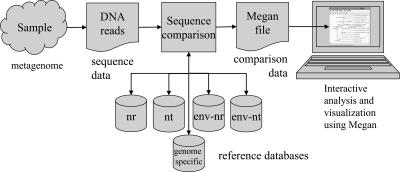
MEGAN is a homology-based taxonomy classificatory that uses metagenomic reads as input and can use protein databases. It lacks the heuristic k-mer strategy; however, it consumes more time, but it is has high accuracy MEGAN requires previous alignment of your reads (Fig. 5). The preferred program and database for the previous step is BLASTX and the NCBI-NR database; MEGAN uses an NCBI taxonomy, the database must be compatible with this taxonomy structure. The results of the alignment are provided to MEGAN, different formats are allowed. MEGAN filters your hits and selects the LCA, which represents the more conserved taxonomic rank of the sequence. Select the LCA improves the precision of the program, but it also reduces the resolution. On the other side, the raw alignment consumes time, but it also improves the catch of sequences. Finally, the MEGAN results can be easily translated into multiple visualization options: Krona, BLASTatla, Strainer, etc.
Kaiju is another homology-based program with metagenome reads as input and protein database as reference. The program obtains the six protein reading frames of the input sequence and search for exact matches into the reference database. Kaiju returns the best hit; if two or more hits have the same score, kaiju assigns the LCA as the taxonomic label of the read.
CARMA is another homology-based classifier, it searches for conserved Pfam domains. The program uses metagenomic reads as input.
MEGAN is a homology-based taxonomy classificatory that uses metagenomic reads as input and can use protein databases. It lacks the heuristic k-mer strategy; however, it has more precision values at higher time/classification rates. MEGAN requires previous alignment of your reads. The preferred program and database for the previous step is BLASTX and the NCBI-NR database; MEGAN uses an NCBI taxonomy, the database must be compatible with this taxonomy structure. The results of the alignment are provided to MEGAN, different formats are allowed. MEGAN filters your hits and selects the LCA, which represents the more conserved taxonomic rank of the sequence. Select the LCA improves the precision of the program, but it also reduces the resolution. On the other side, the raw alignment consumes time, but it also improves the catch of sequences. Finally, the MEGAN results can be easily translated into multiple visualization options: Krona, BLASTatla, Strainer, etc.
Kaiju is another homology-based program with metagenome reads as input and protein database as reference (Fig. 6). The program obtains the six protein reading frames of the input sequence and search for exact matches into the reference database. Kaiju returns the best hit; if two or more hits have the same score, kaiju assigns the LCA as the taxonomic label of the read.
CARMA is another homology-based classifier, it searches for conserved Pfam domains. The program uses metagenomic reads as input.
Fig. 6: Kaiju web server. Kaiju provides pre-built databases. Besides, you can submit jobs with adjustable parameters. Kaiju
The last type of homology-based programs are the marker-gene approaches. The databases of these programs contain marker genes, such genes can be single-copy genes shared across domains, or marker genes of specific clades. Use only a small set of genes reduces the classified percentage of reads, reduces the false positives, and improves the computational speed. “They are meant to be used to characterize the distribution of organisms present in a given sample, rather than labelling every single read.” Another advantage of use the single-copy genes is that it has a better landscape of the real abundance counts. MetaPhlAn and MetaPhyler are some programs that use marker genes to classify the metagenome reads. As curious information, MetaPhlAn was used in at the beginning of the Human microbiome program.
On the other hand, the composition-based programs use machine learning and/or statistical procedures to analyse the reference genomes composition and define shared patterns by each specific clade. Since the composition-based programs find footprints repeated in entire genomes and clades, these algorithms have a wider perspective than the homology classifiers. Besides, the recovery of patterns across clades makes it possible to classify a sequence with only a few representative genomes of the clade of the sequence. For these reasons, the composition-based programs are more suitable to classify complex environments, since the recent databases encompass only a small fraction of the microbial diversity. According to some authors, a small fraction of the reference genomes is enough to recover the clade footprints, which reduces the memory requirements and consumed time. The frequency of short k-mers in a genome are the preferred patters to define a clade.
PhyloPythia is a machine learning algorithm used to classify metagenome contigs. It is based on support vector machine; the program uses variable length k-mer composition. On the other hand, Phymm identifies oligonucleotides shared across a phylogenetic group using interpolated Markov models (IMMs), then, it assigns taxonomy labels to the raw reads of a microbiome sample. PhymmBL is a hybrid approach, it uses composition-based algorithms (Phymm) and homology-based algorithms (BLAST alignments); PhymmBL has shown accuracy improvement.